
Future Directions in Myelofibrosis Therapy: Emerging Novel Agents and New Treatment Strategies
 |
Elizabeth Hexner, MD
Associate Professor of Medicine Hospital of the University of Pennsylvania Medical Director Center for Cellular Immunotherapies Philadelphia, Pennsylvania |
|
Future Directions in Myelofibrosis Therapy: Emerging Novel Agents and New Treatment Strategies
Beyond Ruxolitinib: The Clinical Need for New Therapies for MF
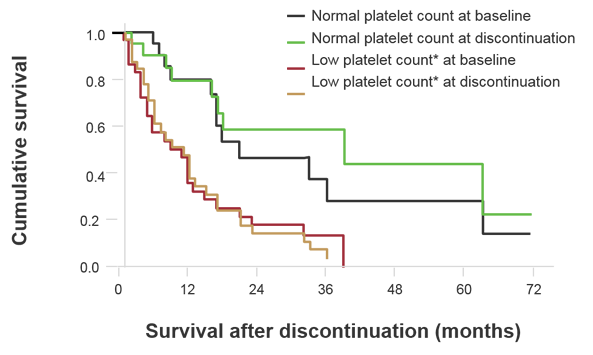
It is clear that survival outcomes beyond ruxolitinib is dismal, as findings from a phase 1/2 study showed that among 86 patients who discontinued ruxolitinib, 30 died while taking the agent, and the median survival of 56 patients was 14 months after discontinuation (Figure 1).1 Low platelet count at baseline was associated with shorter survival, but this may also be due to the fact that this study included a population at high risk for death.
Figure 1. Survival Following Ruxolitinib Treatment2
Ruxolitinib: Identifying and Managing Progression or Resist
The features of progression and treatment options are presented in Table 1. Although an increase in spleen size is an indicator of progression, caution is warranted when determining that the spleen has indeed become larger since our measurements tend to be rather imprecise, even when documented in the patient’s medical record. Although I would recommend physicians obtain baseline data including spleen size prior to initiating therapy, I would caution against declaring that a patient is failing therapy if the spleen size seems to be larger but they are otherwise feeling well. Symptoms are also a good marker of progression, and these also can be captured at baseline and followed during therapy. This can be obtained using tools such as the MPN 10 Symptom Form. Cytopenias are recognized harbingers of progression, but it is important to remember that this also could be a side effect of ruxolitinib. Emerging leukocytosis can be indicative of accelerated and blast phase disease, and in situations where this significantly impacts quality of life, I would recommend a hypomethylating agent in combination with ruxolitinib.
Table 1. Identification and Management of Progression/Resistance on Ruxolitinib*
Feature |
Treatment Options |
Spleen |
|
Symptoms |
|
More anemia or thrombocytopenia |
|
Leukocytosis |
|
Blasts |
|
*Adapted from Gupta V, Cerquozzi S, Foltz L, et al. Patterns of ruxolitinib failure and its management in myelofibrosis: Perspectives of the Canadian Myeloproliferative neoplasm Group. JCO Oncol Pract. 2020 Mar 5:JOP1900506. doi: 10.1200/JOP.19.00506. Online ahead of print
Defining Ruxolitinib Failure
According to the International Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) 2013 response criteria, progressive disease is defined as2:
- Appearance of a new splenomegaly that is palpable at least 5 cm below the left costal margin (LCM) or
- A ≥100% palpable increase in palpable distance, below LCM, for baseline splenomegaly of 5 to 10 cm or
- A 50% increase in palpable distance, below LCM, for baseline splenomegaly of >10 cm or
- Leukemic transformation confirmed by a bone marrow blast count of ≥20% or
- A peripheral blood blast content ≥20% associated with absolute blast count of ≥1 x 10/L that lasts at least 2 weeks
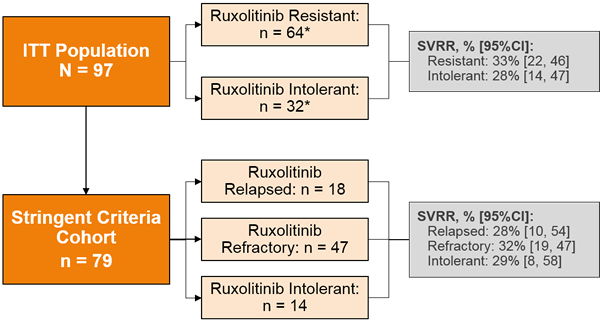
However, recent clinical trials have employed a stringent definition of ruxolitinib failure (Figure 2),3,4 such as in a re-analysis of the JAKARTA-2 trial.3 Figure 3 compares the results of the original intention-to-treat (ITT) analysis and the results of the more stringent cohort in this trial.3
Figure 2. Definitions of Ruxolitinib Failure in Recent Clinical Trials3,4

Bl=baseline; RBC=red blood cell; RUX=ruxolitinib; SVR=spleen volume response; Tx=therapy
aHarrison CN, Schaap N, Vannuchi AM, et al. Fedratinib (FEDR) in myelofibrosis (MF) patients previously treated with ruxolitinib (RUX): A reanalysis of the JAKARTA-2 study. ASCO 2019. Abstract 7057. bDaver N, Kremyanskaya M, O'Connell C, et al. A phase 2 study of the safety and efficacy of INCB050465, a selective PI3K inhibitor, in combination with ruxolitinib in patients with myelofibrosis. ASH 2018. Abstract 353
Figure 3. Spleen Volume Response Rates by Prior Ruxolitinib Outcomes: ITT Population and Stringent Criteria Cohort3
Optimizing Ruxolitinib-based Therapy: Novel Agents and Approaches for MF
Table 2 presents the current clinical trials investigating combination ruxolitinib approaches, which are mainly in phase 1/2 testing. This section will highlight the most compelling data from recent key studies.
Table 2. Ongoing Clinical Trials in Ruxolitinib-based Combination Therapies
Adapted from Economides MP, Verstovsek S, Pemmaraju N. Novel therapies in myeloproliferative neoplasms: Beyond JAK inhibitors. Curr Hematol Malig Rep. 2019;14:1440-1468.
Imetelstat
Imetelstat is a covalently lipidated 13-mer oligonucleotide that binds to human telomerase RNA and has been shown to be a potent competitive inhibitor of telomerase activity.5,6 The phase 2 IMBARK trial was designed to test the agent at two dose levels, 4.7 mg/kg q 3 weeks (n=48) and 9.4 mg/kg q 3 weeks (n=59), in patients with intermediate- or high-risk MF that progressed during or after JAK-inhibitor therapy and who had active symptoms of disease.7 The median time on JAK inhibitor therapy was 23 months, and the median platelet count was 147 x 109/L. About a quarter of patients had triple-negative disease (ie, no mutations of JAK2, MPL, or CALR), and 67% had high-molecular risk disease (≥1 mutation of ASXL1, EZH2, SRSF2, or IDH1/2).
At clinical cut-off, the median follow-up was about 23 months, and the median duration of treatment was 6 months.7 At week 24, 6 patients in the 9.4 mg/kg arm had a ≥35% spleen volume response (SVR) versus 0 in the 4.7 mg/kg group; moreover, 23 patients at the higher dose achieved ≥10% SVR. More patients receiving the higher dose also had higher total symptom score (TSS) reduction at week 24 versus those on the lower dose (19 vs 3 patients, respectively). Importantly, median overall survival (OS) was about 30 months in the 9.4 mg/kg arm versus 20 months in the 4.7 mg/kg arm, suggesting a signal of improved survival with imetelstat.
A case-control study evaluated imetelstat versus best available therapy (BAT) in a cohort of patients similar to those included in the IMBARK study from Moffitt Cancer Center.8 Data showed that imetelstat conferred a 65%-67% lower risk of death versus BAT, supporting the previous findings that the agent provides favorable OS compared to historical real-world findings.
Navitoclax
Navitoclax is a novel small molecule that binds with high affinity to BCL-XL, BCL-2, and BCL-W, causing cell death by apoptosis,9 and has demonstrated cytotoxic activity in myeloproliferative neoplasm (MPN)-derived cell lines.10-13 This agent has was investigated in a phase 2, single-arm open-label trial in combination with ruxolitinib in patients with primary or secondary MF.14 The trial was based on the hypothesis that the addition of navitoclax to ruxolitinib would overcome resistance to JAK2 inhibitors. Patients had to have received at least 12 weeks of continuous ruxolitinib therapy before starting navitoclax, which was given at a starting dose of 50 mg once daily. Dose-escalation of navitoclax was allowed based on platelet count and tolerability to a maximum dose of 300 mg.
Results showed that 23 of the 34 patients enrolled on the study achieved the 300 mg dose of navitoclax, and 22 of 25 patients who enrolled on ruxolitinib at doses of >10 mg BID had ruxolitinib dose reduced to 10 mg BID.14 About a third of the 24 evaluable patients had a ≥35% reduction in spleen volume from baseline, and navitoclax also elicited a 20% improvement in TSS. Reductions in driver mutation allelic burden of >5% also were reported, as well as improvements in bone marrow fibrosis.
All patients experienced an adverse event of any grade, and 8 patients had a serious adverse event, which included anemia, pancytopenia, splenic infarction, upper abdominal pain, vomiting, chest pain, pneumonia, and abnormal liver function.14 One patient experienced grade 5 pneumonia, but this was unrelated to navitoclax therapy.
Luspatercept
This agent is a first-in-class erythroid maturation agent that binds several TGF-β superfamily ligands to diminish Smad2/3 signaling and enhance late-stage erythropoiesis15 that is approved in the United States for use in adult patients with β-thalassemia who require regular red blood cell (RBC) transfusions.16 On April 3, 2020, the agent was approved for use in for the treatment of anemia failing an erythropoiesis-stimulating agent and requiring two or more RBC units over 8 weeks in adult patients with very low- to intermediate-risk myelodysplastic syndromes (MDS) with ring sideroblasts or with MDS/MPN with ring sideroblasts and thrombocytosis.17 The drug is also being studied in an open-label phase 2 trial in MDS patients who were anemic and transfusion independent, and in those who were transfusion dependent (Figure 4).18 Luspatercept was given at a dose of 1.0 mg/kg with titration up to 1.75 mg/kg every 21 days for 168 days, with or without ruxolitinib.
Figure 4. Study Design of an Open-label Phase 2 Trial of Luspatercept in Myelofibrosis18
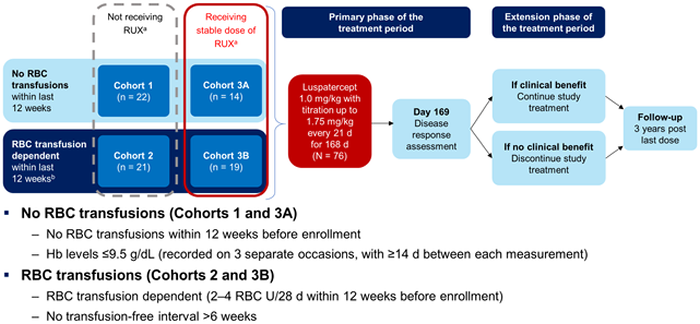
D=days; Hb=hemoglobin; RBC=red blood cell; RUX=ruxolitinib
Results showed that the patients receiving the combination regimen derived greater benefit on both the primary and secondary endpoints.18 The median duration of RBC independence of 12 weeks or greater was 32 and 39 weeks for cohorts 2 and 3B, respectively.
CPI-0610
This agent is a small molecule inhibitor of the bromodomain and extraterminal family of proteins that is being evaluated in the phase 2 MANIFEST trial as both monotherapy and in combination with ruxolitinib (Figure 5).19 The primary endpoint for transfusion-dependent patients was conversion from transfusion dependency to transfusion independence, defined as the absence of RBC transfusions over any 12 weeks. For non-transfusion dependent patients, the primary endpoint was SVR of ≥35% from baseline by magnetic resonance imaging or computed tomography at week 24. For both cohorts, the key secondary endpoint was a reduction of ≥50% in TSS as measured by the Myelofibrosis Symptom Assessment Form at week 24.
Figure 5. Study Design of the MANIFEST Trial19
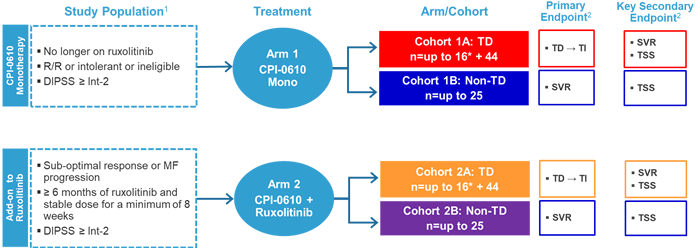
1ClinicalTrials.gov identifier NCT02158858. 2Other endpoints: anemic response, RBC transfusion rate, safety, pharmacokinetics, proinflammatory cytokine levels, bone marrow morphology, and mutant allele burden.
*Will follow Simon 2-stage design.
DIPSS=Dynamic International Prognostic Scoring System; MF=myelofibrosis; mono=monotherapy; SVR=spleen volume response; TD=transfusion dependent; TI=transfusion independent; TSS=total symptom score
Among transfusion-dependent patients who received the combination therapy (Cohort 2A), SVR 35 at week 24 was 25%, which was a median change from baseline of -24.9%.19 At the same time point, TSS ≥50% was 54%, which was a median change from baseline of -58.8%. Moreover, 6 of 14 patients became transfusion independent. Among patients who were not transfusion dependent, CPI-0610 monotherapy provided a greater hemoglobin benefit versus those receiving the combination therapy (55% vs 13%). CPI-0610 was generally well tolerated as monotherapy or as add-on therapy with ruxolitinib; the most common hematologic adverse event was thrombocytopenia. Grade ≥3 thrombocytopenia was reported in 9 patients, which were managed with dose interruption or reduction.
PRM-151
Pentraxin-2 (PTX-2) is an endogenous regulator of tissue repair that binds damaged tissue and monocytes and macrophages.20 In patients with MF, PTX-2 are low. PRM-151 is a recombinant human PTX-2 that has been shown to inhibit primary myelofibrosis (PMF) bone marrow-derived fibrocyte differentiation in vitro, slow development of bone marrow fibrosis , and significantly improve survival in mouse models.21 This agent was studied in a randomized phase 2 study in patients with MF previously treated with ruxolitinib.22 Patients were randomly assigned in a 1:1:1 fashion to receive PRM-151 IV at 10 mg/kg, 3 mg/kg, or 0.3 mg/kg; on days 1, 3, and 5, and then every 28 days for 9 cycles. Results showed that PRM-151 improved bone marrow fibrosis, and responses were similar among all treatment arms. Reduction of ≥1 fibrosis grade was observed after as few as 3 cycles of the agent.
Creating Meaningful Physician-Patient Communications to Optimize Outcomes
Recent data show that there is a lack of alignment between physician and patient perceptions regarding MF management, which can impair determination of the optimal treatment regimen for the patient.23 It is thus critical to improve physician-patient communication to ensure that the patient’s goals of therapy are included when making treatment decisions.
The MPN Landmark Survey was designed to evaluate gaps between patient perceptions of their disease management and physician self-reported practices.23 The study included 813 patient respondents with MPNs and 457 physician hematologists/oncologists in the United States. Although about two-thirds of patients were satisfied with physician management and communication regarding MF, results regarding the physician versus patient perception of communication about MF symptoms revealed discrepancies between discussion of symptoms and prioritizing those symptoms (Table 3).23
Table 3. Patient and Physician Respondent Perspectives on Communication About MF Symptoms in the MPN Landmark Survey23
MF Respondents |
||
| Variable, n (%) | Patients n = 207 |
Physicians n = 156 |
| Physician symptom assessment | ||
| Proactively asks how the patient is feeling | 105 (51) | 55 (35) |
| Waits for the patient to mention symptoms | 44 (21) | 8 (5) |
| Asks about the patient’s most important symptoms | 41 (20) | 86 (55) |
| Physician uninterested or does not ask about symptoms | 10 (5) | NA |
| Fills out a symptoms list/review each symptom | 7 (3) | 6 (4) |
| Other | NA | 1 (1) |
| Physician communication of potential symptoms and disease progression | ||
| Only discusses current symptoms | 89 (43) | NA |
| Asks about a full and comprehensive list of symptoms | 73 (35) | 81 (52) |
| Outlines most likely symptoms | 19 (9) | 43 (28) |
Findings also showed that many patients (about less than half in most instances) were unable to recognize the common symptoms of MF, including difficulty sleeping, numbness/tingling in the hands and feet, and dizziness/vertigo/lightheadedness.23 Moreover, the survey results showed notable differences between physicians’ and patients’ goals of treatment (Figure 6).23 For patients, the most important goal was to slow or delay disease progression, whereas for physicians, this was to improve symptoms.
Figure 6. Most Important Treatment Goals for Myelofibrosis from Patient-Physician Respondent Comparisons in the MPN Landmark Survey23
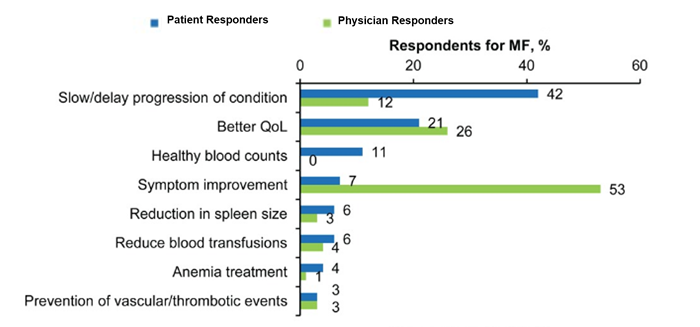
MF=myelofibrosis; QoL=quality of life
As mentioned above, it is important for physicians to utilize the MPN 10 Questionnaire not only before initiating therapy, but also throughout the course of therapy. In this way, physicians can monitor the patient’s MF-related symptoms and the emergence of new ones, which can facilitate and improve communication.