
|
Diagnosis and Classification of PMF
The World Health Organization (WHO) categorizes PMF as either early or prefibrotic stage; or overt, or fibrotic stage, which includes patients with PV or ET that progresses to PMF (post-PV and post-ET, respectively).9 The diagnostic criteria for early and overt PMF are presented in Table 1. The differentiation of early PMF vs ET was based on differences in both their characteristic morphological bone marrow (BM) features and also their characteristic clinical behavior in terms of mortality, venous thrombotic complications, progression to overt PMF, and blast transformation. Compared with ET, early PMF is a less benign disease, with a distinct clinical pattern of progression, with also an increased bleeding tendency. However, although early PMF can transform into overt PMF, Barbui and colleagues9 showed that the transformation risk is 15% after 15 years of follow up, and the transformation rate to acute myeloid leukemia (AML) is 10% after 15 years of follow up. Early PMF can thus be considered a benign disease compared with overt PMF and can be treated similarly as ET.
Table 1.9 | ||
Primary myelofibrosis (PMF) a |
||
Prefibrotic/early PMF (pre-PMF) |
Overt PMF |
|
Major criteria |
||
1 |
Megakaryocytic proliferation and atypiab, without reticulin fibrosis > grade 1c, accompanied by increased age-adjusted BM cellularity, granulocytic proliferation and often decreased erythropoiesis |
Megakaryocyte proliferation and atypiab accompanied by either reticulin and/or collagen fibrosis (grade 2 or 3) |
2 |
Not meeting WHO criteria for BCR-ABL1 + CML, PV, ET, MDS, or other myeloid neoplasm |
Not meeting WHO criteria for BCR-ABL1 + CML, PV, ET, MDS or other myeloid neoplasm |
3 |
Presence of JAK2, CALR, or MPL mutation or in the absence of these mutations, presence of another clonal markerd or absence of minor reactive BM reticulin fibrosise |
Presence of JAK2, CALR, or MPL mutation or in the absence, the presence of another clonal markerd or absence of evidence for reactive BM fibrosisf |
Minor criteria |
||
1 |
Presence of one or more of the following, confirmed in two consecutive determinations: |
Presence of one or more of the following confirmed in two consecutive determinations: |
• Anemia not attributed to a comorbid condition |
• Anemia not attributed to a comorbid condition |
|
• Leukocytosis ≥ 11 × 109/L |
• Leukocytosis ≥ 11 × 109/L |
|
• Palpable splenomegaly |
• Palpable splenomegaly |
|
• LDH level above the upper limit of the institutional reference range |
• LDH level above the upper limit of the institutional reference range |
|
• Leukoerythroblastosis |
||
Table adapted from Barbui T, et al. Blood Cancer J. 2015;5:e337. and Arber DA, et al. Blood.2016;127:2391–2405
BM=bone marrow; CML=chronic myeloid leukemia; MDS=myelodysplastic syndrome; LDH=serum lactate dehydrogenase
a Diagnosis of prefibrotic/early PMF requires all three major criteria and at least one minor criterion. Diagnosis of overt PMF requires meeting all three major criteria and at least one minor criterion
b Small-to-large megakaryocytes with aberrant nuclear/cytoplasmic ratio and hyperchromatic and irregularly folded nuclei and dense clustering
c In cases with grade 1 reticulin fibrosis, the megakaryocyte changes must be accompanied by increased BM cellularity, granulocytic proliferation, and often decreased erythropoiesis (that is, pre-PMF)
d In the absence of any of the three major clonal mutations, the search for the most frequent accompanying mutations (ASXL1, EZH2, TET2, IDH1/IDH2, SRSF2, SF3B1) are of help in determining the clonal nature of the disease
e Minor (grade 1) reticulin fibrosis secondary to infection, autoimmune disorder or other chronic inflammatory conditions, hairy cell leukemia or other lymphoid neoplasm, metastatic malignancy, or toxic (chronic) myelopathies
f BM fibrosis secondary to infection, autoimmune disorder, or other chronic inflammatory conditions, hairy cell leukemia, or other lymphoid neoplasm, metastatic malignancy or toxic (chronic) myelopathies
As shown in Figure 1, overt PMF is characterized by progressive organomegaly and extramedullary hematopoiesis, progressive cytopenias, and progressive constitutional symptoms.6 Patients with overt PMF have a substantially decreased quality of life (QoL); progressive incapacitation and immobility; and increased risks for disease-related complications; leukemic transformation, and premature death.
| Figure 1.6 |
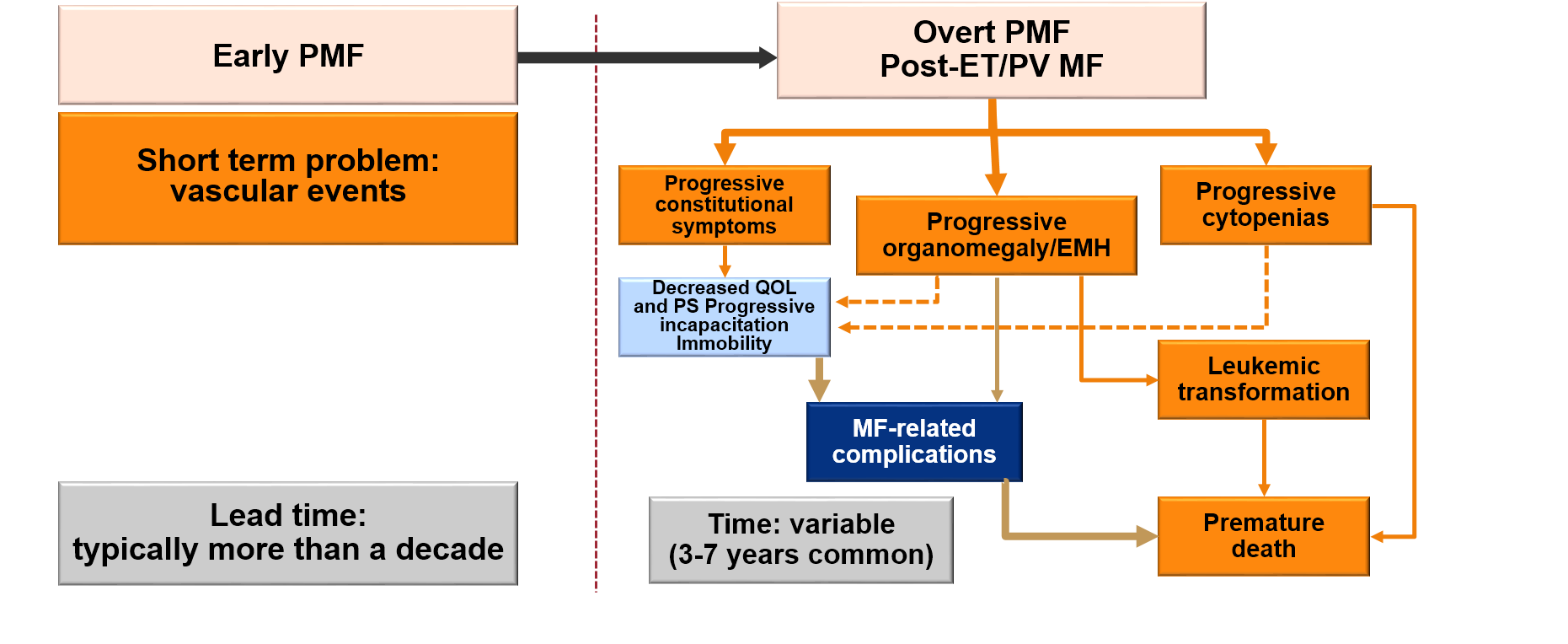 |
| EMH=extramedullary hematopoiesis; ET=essential thrombocythemia; PMF=primary myelofibrosis; PS=performance status; PV=polycythemia vera; QOL=quality of life. |
Pathobiology of MF
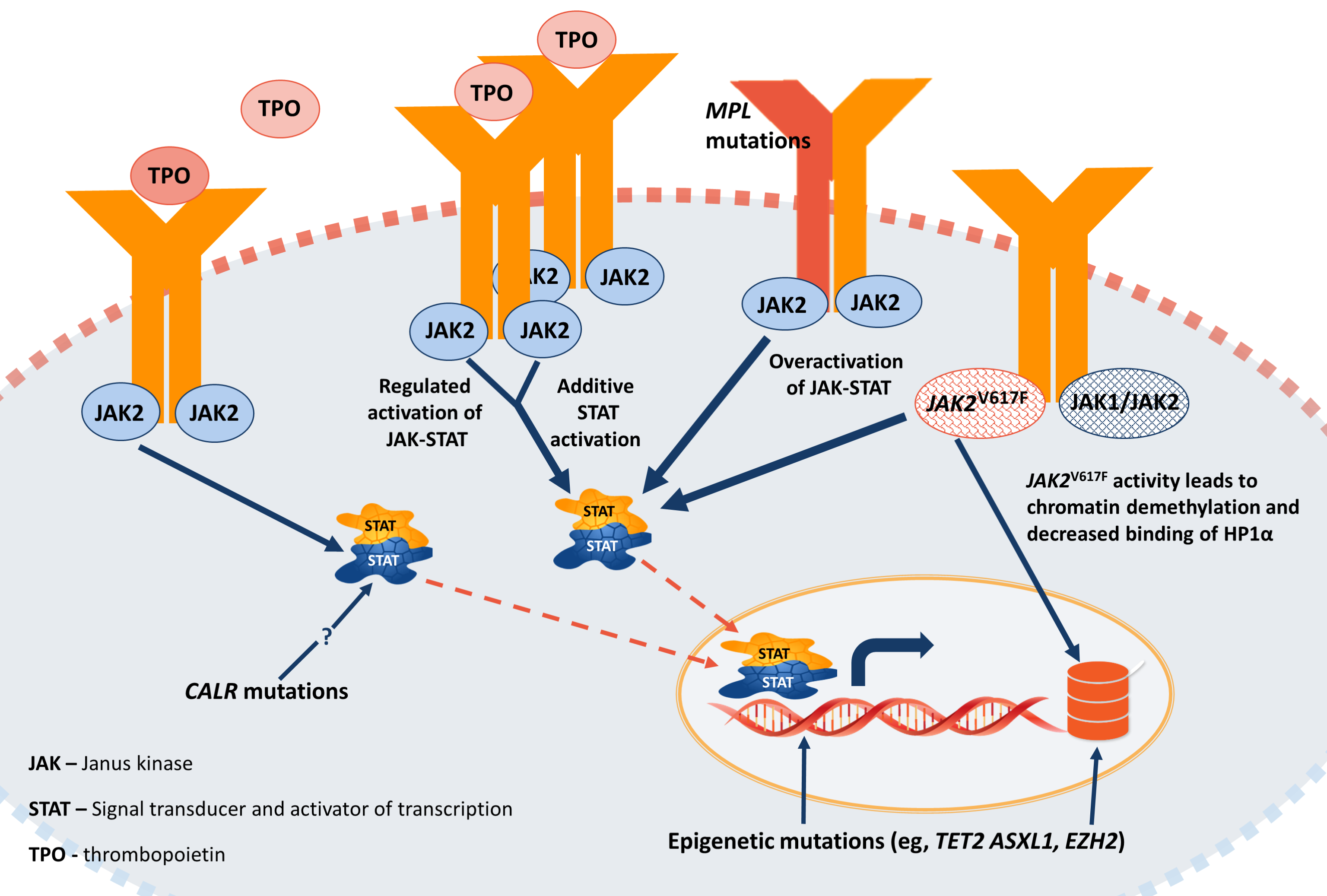
Three oncogenic driver mutations in MF have been identified: JAK2, CALR, and MPL, all which activate JAK-STAT signaling, which ultimately drives MF (Figure 2).10
These mutations are essentially mutually exclusive and have been associated with specific prognoses. For example, CALR mutations occur in 27% of patients with MF and is associated with a decreased risk of thrombosis and thus confers a favorable prognosis of about 18 years.11 JAK2 and MPL mutations are detected in 65% and 4% of patients, respectively, and are each associated with a prognosis of about nine years. Triple-negative disease occurs in about 8.6% of patients and has the worst prognosis of three years.
Non-driver mutations of high molecular risk have also been identified, such as IDH, EZH2, ASXL1, and SRSF2.12-14 The presence of two or more of these mutations worsens survival in MF, and may even negatively impact the presence of a favorable driver mutation, which has been observed when both CALR and ASXL1 mutations were present.
| Figure 2.10 |
 |
Classical Prognostic Models of Myelofibrosis
The so-called classic models—the IPSS, the DIPSS, and DIPSS Plus—include age, constitutional symptoms, hemoglobin level, leukocyte count, and peripheral blood blasts.4,5,15 However, only the DIPSS-Plus model includes other parameters such as unfavorable karyotype, red blood cell (RBC) transfusion dependence, and platelet count.5 Moreover, the IPSS can be used only at diagnosis, whereas the DIPSS and DIPSS-Plus may be used at any timepoint during the course of disease.4,5,15 There is some debate regarding which prognostic model should be used at specific timepoints, and which one is the best. Findings from a Spanish study that employed all three models showed that patients moved from one category to another during the same timepoint when a different model was used.16 It is thus recommended to choose the model based on the individual patient’s available clinical information. Finally, it is important to note that these models are intended to determine a patient’s life expectancy with a primary reason to identify those patients who would have life expectancy shorter than five years, in which case autologous stem cell transplant (ASCT) should be considered.
Newer Prognostic Models: MIPSS70 and MIPSS70-Plus
As mentioned, the discovery of relevant mutations in MF continues to shape the development of prognostic models. The MIPSS70 and MISS70-Plus prognostic tools integrate these mutational data with the individual patient and disease characteristics (Figure 3).17,18 Due to the complexity of these tools and to facilitate determination of a patient’s prognostic score, both are available online at http://www.mipss70score.it/.
Figure 3.18
Key Elements of the MIPSS70 and MIPSS70-Plus
- Hb <10 g/dL*
- WBC >25 x 109/L
- PLT <100 x 109/L
- Blasts ≥2%*
- Fibrosis > grade 1
- Constitutional symptoms*
- Absence of type 1/-like CALR mutation*
- HMR mutations*
- ASXL1
- EZH2
- SRSF2
- IDH1/2
- Two or more HMR*
*Unfavorable karyotype
Treatment Selection for MF: The Role of JAK Inhibitors
Planning management regimens for MF should include not only the patient’s prognostic score but also the specific clinical need for therapy. Objectivizing the patient’s quality of life is also important, which should be assessed at the initiation of therapy as well as during the treatment course.19 Current guidelines recommend the use of questionnaires such as the MPN10 in daily practice.
Of the currently available therapies, JAK inhibitors are the most effective to improve symptomatic splenomegaly and constitutional symptoms/QoL. Ruxolitinib was also shown to possibly prolong life of MF patients.
Ruxolitinib
Ruxolitinib is an oral selective tyrosine kinase inhibitor (TKI) of JAK 1/2, was approved in 2011 for the treatment of patients with intermediate- or high-risk MF, including PMF, post-PV, and post-ET. Approval of this agent was based on combined results from two phase 3 studies, the Controlled Myelofibrosis Study with Oral JAK Inhibitor Treatment (COMFORT)-I and -II trials.
Both trials included patients with intermediate-2 and high-risk MF, with platelet counts of ≥100 x 10-9/L and splenomegaly.20,21 Patients received either ruxolitinib or placebo in the double-blind, randomized COMFORT-I trial, and ruxolitinib or best available therapy (BAT) in the open-label COMFORT-II. The primary endpoint was a ≥35% reduction in total spleen volume (as assessed by MRI or CT) from baseline to week 24 in COMFORT-I, and from baseline to week 48 in COMFORT-II. In the initial analyses of these studies, this was achieved by 41.9% of patients in the ruxolitinib group vs .07% of those on placebo in COMFORT-I, and by 28% on ruxolitinib and 8% on BAT (most commonly, hydroxyurea in 47%) in COMFORT-II. Ruxolitinib yielded some degree of reduction in spleen volume in 97% of patients overall in both trials, and long-term use of this agent has been associated with substantial and durable responses and survival benefit.
Findings from an exploratory analysis of five-year data pooled from 528 patients in the COMFORT trials showed the continued efficacy of ruxolitinib.22 For patients who received ruxolitinib, the risk of death was reduced by 30% compared with those in the comparator groups (median OS, 5.3 vs 3.8 years, respectively; P=.0065). After correcting for crossover, this OS benefit was especially more evident among patients who were originally randomized to receive ruxolitinib vs those who crossed over from the control group (median OS, 5.3 vs 2.3 years). Ruxolitinib was also observed to extend OS vs controls in an analysis of OS censoring patients at the time of crossover (median OS, 5.3 vs 2.4 years, respectively; P=.0013). Finally, ruxolitinib provided an OS benefit regardless of anemia status at baseline or transfusion requirements at week 24.
The most common nonhematologic toxicities reported in the COMFORT studies were fatigue, diarrhea, and asthenia, most of which were grade 1 or 2 (Table 2).20,21,23 The most common hematologic adverse effects were anemia and thrombocytopenia, most of which were grade 3 and 4. Cytopenias were treated with dose modifications or interruptions, and red blood cell infusions for anemia. In both trials, mean hemoglobin levels decreased initially, but recovered after 8 to 12 weeks of treatment. In clinical practice, it is possible to manage the emergence of anemia with medication and not decrease the ruxolitinib dose.
| Table 2.20,21,23 | ||||||||||||||||||||||||
| Hematologic adverse eventsa in the phase III COMFORT trials, regardless of relation to study drug | ||||||||||||||||||||||||
| ||||||||||||||||||||||||
| BAT best available therapy, COMFORT COntrolled MyeloFibrosis study with ORal JAK inhibitor Treatment, NR not reported aNew or worsening hematologic events based on laboratory values | ||||||||||||||||||||||||
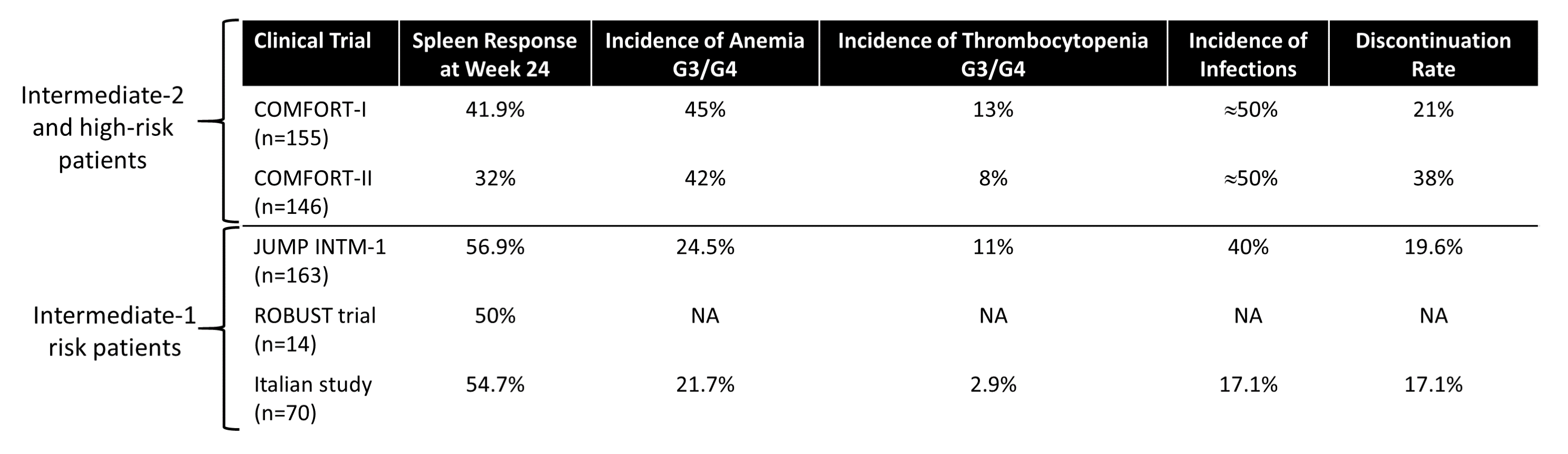
Other data indicate that early intervention with ruxolitinib may provide higher response rates with lower toxicities vs in higher-risk disease (Table 3).24-27 This may be due to better bone marrow reserves in early disease (less baseline anemia and thrombocytopenia), which allows patients to receive therapy with less toxicity and higher ruxolitinib dose.
| Table 3.24-27 |
 |
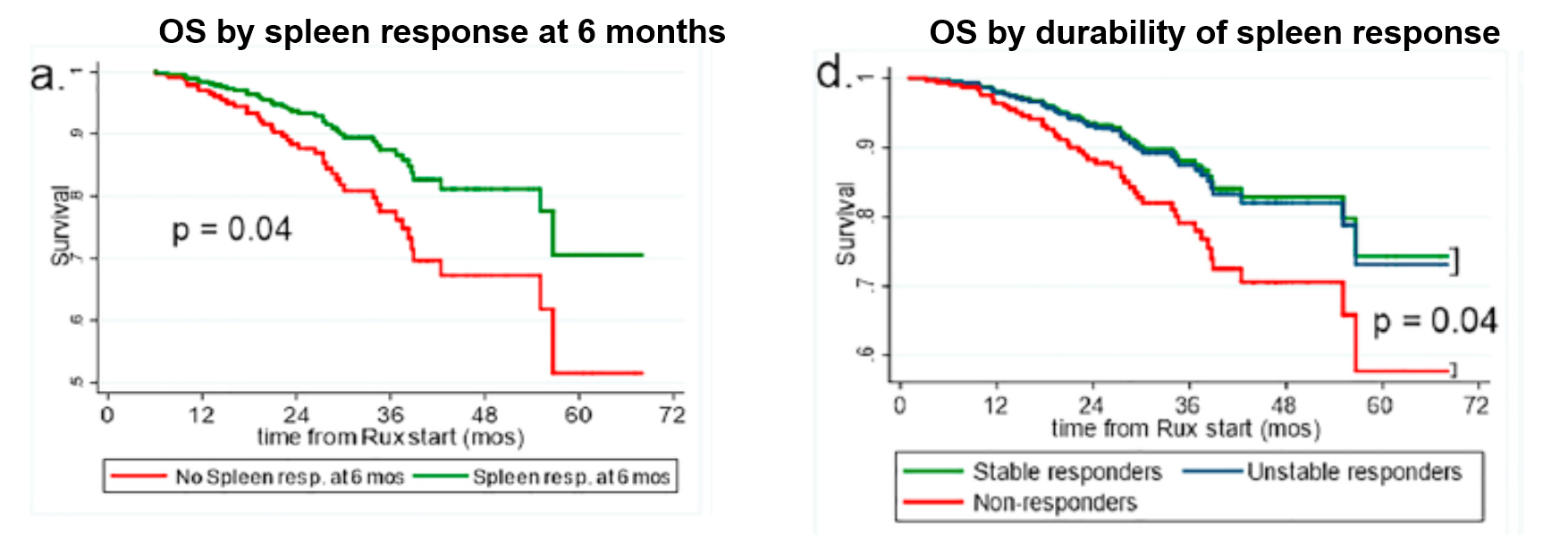
The correlation of OS to spleen response to ruxolitinib has been shown in multiple studies.28-30 Ruxolitinib provides improvement in OS within six months of therapy, and this benefit correlates with the durability of spleen response (Figure 4). Moreover, there is also a correlation between dose and improvements in spleen volume and total symptom score (TSS).31 Ruxolitinib at doses lower than 10 mg BID is not effective in the long term. It is thus recommended that if a patient must start on a dose >10 mg BID, it should be escalated quickly to the maximum safe dose to ensure optimal outcomes.
| Figure 4.26,29 |
 |
A correlation of mutational profile and response to ruxolitinib also has been described.32 Findings from one study showed that splenic response was not associated with JAK2, CALR, MPL, or TN mutation status. However, patients with one or more mutations in ASXL1, EZH2, or IDH1/2 and those with ≥3 mutations of any type were significantly less likely to have a spleen response than those with no ASXL1, EZH2, or IDH1/2 mutations (52% vs 80%; P=.01) and those with ≤2 mutations (25% vs 60.5%; P=.001), respectively.
For patients who discontinue ruxolitinib, survival outcomes are poor, with a median OS of 14 months in one study.33 Shorter survival has been observed in patients with low platelets at the start of therapy (Plt <260 x 109/L; HR=2.7, P=.006) and at the end of therapy (Plt <100 x 109/L; HR=4.1, P=.001). Clonal evolution has contributes to significantly shorter survival after ruxolitinib therapy (6 vs 16 months in patients without clonal evolution, P=.006).32 In one study, 35% of patients acquired a new mutation while receiving ruxolitinib, of which 61% were mutations in ASXL1. Most patients require salvage therapy following ruxolitinib discontinuation, which can include allogeneic transplantation, lenalidomide, thalidomide, hydroxyurea, interferon, or danazol; or rechallenge with ruxolitinib,34 but only about a quarter of patients respond to treatment.35 As such, there is an ongoing clinical need to develop new therapeutic approaches for patients following ruxolitinib discontinuation.
Fedratinib
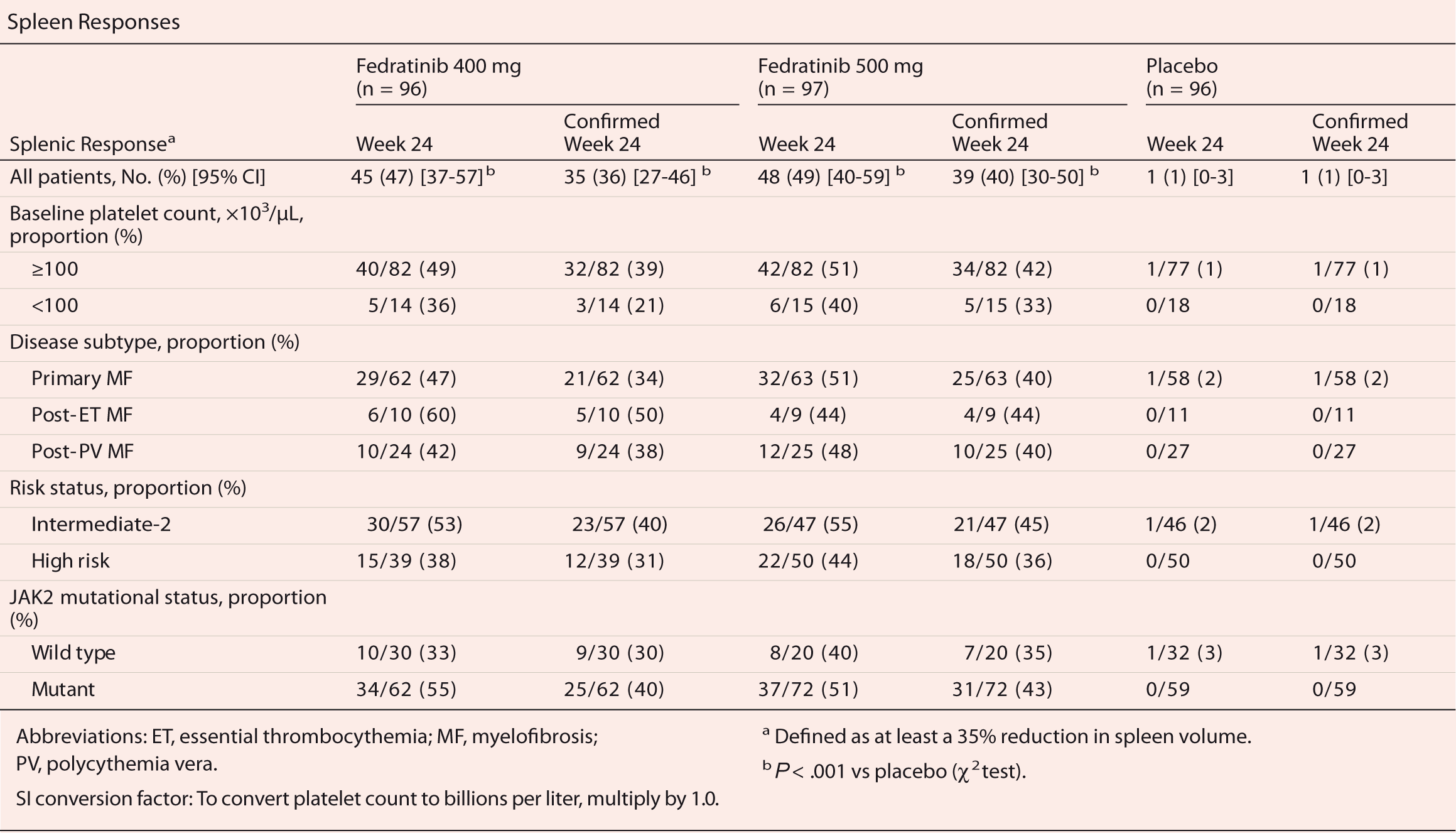
Fedratinib is an oral JAK2/FLT3 TKI that was approved in 2019 for treatment of patients with intermediate-2 or high-risk MF, including PMF, post-PV, and post-ET, based on findings from the phase 3 JAKARTA trial.36 In this study, patients (N=298) were randomly assigned to receive fedratinib at either the 400 mg (n=96) or 500 mg dose (n=97); or placebo (n=96), for at least six consecutive 4-week cycles. The primary endpoint was splenic response (≥35% reduction in spleen volume, as assessed by MRI or CT) at week 24, and confirmed four weeks later. The main secondary endpoint was symptom response, defined as a ≥50% reduction in total symptom score (TSS), as assessed by the modified Myelofibrosis Symptom Assessment Form.
Result showed that at both doses, fedratinib significantly reduced spleen volume vs placebo at 24 weeks and confirmed four weeks later.36 Thirty-six of patients in the fedratinib 400 mg group and 40% in the 500 mg group met the primary endpoint, compared with 1% of those who received placebo (P<.001 for comparisons of both doses vs placebo (Table 4). In addition to these patients, an additional five patients in each fedratinib treatment arm achieved splenic responses of at least 35% at 24 weeks but were not confirmed four weeks later. JAK2 mutational status, MF disease subtype, and risk status did not affect spleen responses.
| Table 4.36 |
 |
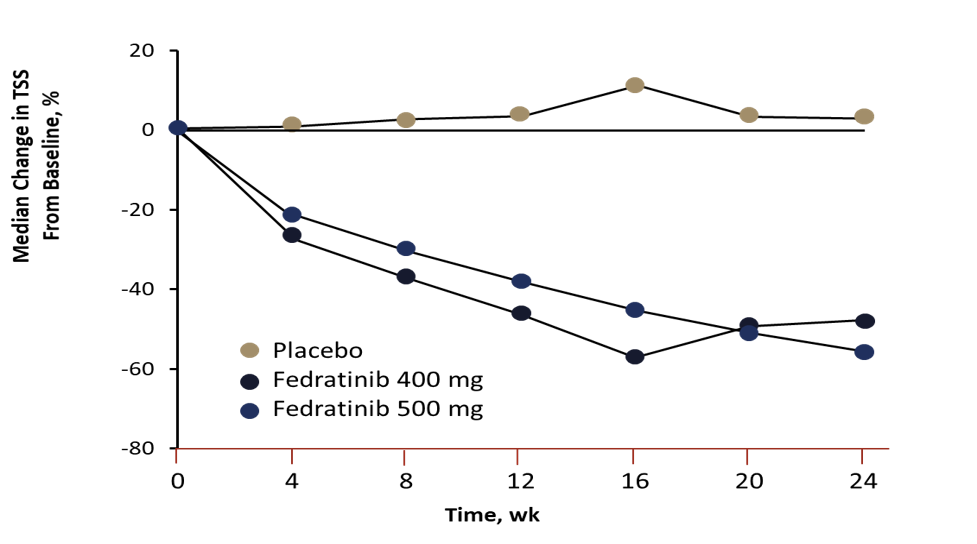
Fedratinib at both doses was also superior to placebo on the secondary endpoint (Figure 5). Thirty-six percent and 34% of patients in the 400 mg and 500 mg groups, respectively, had a reduction of ≥50% in TSS at the 24-week analysis timepoint vs 7% of those in the placebo group.
| Figure 5.36 |
 |
The safety profile of the JAKARTA trial is presented in Table 5.36
Fedratinib was associated with decreased hemoglobin levels, which were lowest after 12 to 16 weeks of treatment, and were partially recovered with the 400 mg dose but not the 500 mg dose.
| Table 5.36 |
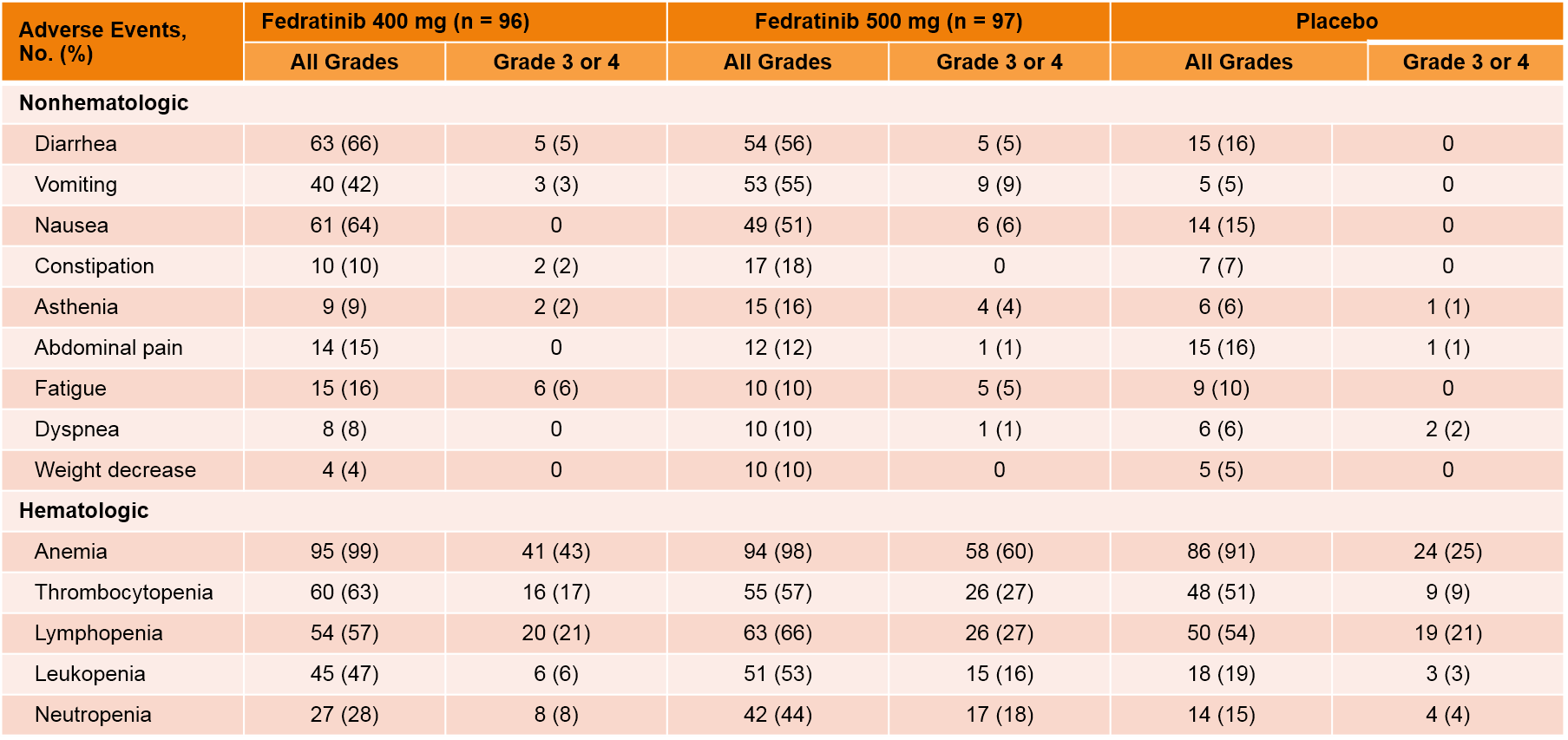 |
One case of Wernicke encephalopathy (WE) and one case of encephalopathy of unknown origin were reported during the study.36 Two other cases of WE were reported after database lock. All cases were women who received fedratinib at the 500 mg dose. Symptoms developed 6 to 44 weeks after the initiation of fedratinib treatment, and pharmacokinetic evaluation of two patients showed above-mean fedratinib levels vs the safety population. Intravenous thiamine was given at diagnosis of encephalopathy, to which most patients responded, but all patients still have some persistent cognitive defects.
The open-label phase 2 JAKARTA-2 study was designed to evaluate fedratinib in the second-line setting after ruxolitinib failure.37 The study included patients with intermediate-2 or high-risk MF (N=97) who were resistant or intolerant to ruxolitinib after at least 14 days of treatment. Fedratinib 400 mg was given once daily, for six consecutive 4-week cycles. The primary endpoint was spleen response (≥35% reduction in spleen volume as determined by CT and MRI). The primary analysis included patients in the per-protocol population, defines as those with spleen volume measurement at baseline and at least one post-baseline. The safety population included all patients who received at least one dose of fedratinib. Fifty-five percent of the 83 assessable patients met the primary endpoint, and 18 discontinued therapy. The most common grade 3-4 AEs were anemia and thrombocytopenia, and there were no reports of encephalopathy.
A recent re-analysis of JAKARTA-2 was conducted to include the intent-to-treat population with new, more stringent definitions of ruxolitinib resistance and intolerance.38 In this analysis, 79 of the 97 enrolled patients met the new criteria for ruxolitinib resistance (n=62) and intolerance (n=14). As in the original analysis, clinically meaningful reductions in splenomegaly and symptom burden were observed. The splenic response rate was 30%, and symptoms response rate was 27%. Toxicities were consistent with prior reports, with no new safety signals identified. In addition, recent findings indicate that fedratinib elicited robust spleen response rates, regardless of the reason for ruxolitinib discontinuation.39,40 An analysis of HRQoL benefit showed that fedratinib provided clinically meaningful improvement across patients subgroups.41